.png?width=300&name=ANAA%20banner%20(1920%20x%201080%20px).png)
Anti-Nephrin Autoantibodies Research
ABOUT THE STUDY
An international study using a novel assay technique reliably detected anti-nephrin autoantibodies in up to 90% of idiopathic nephrotic syndrome. This new research, presented May 25 at the 61st ERA Congress with simultaneous publication in the New England Journal of Medicine, was based on serum/plasma analysis of samples from a multi-center cohort of 357 adults with various glomerular diseases and 192 children with idiopathic nephrotic syndrome, plus 117 healthy controls. The study was led by a group from the University of Hamburg in a collaborative effort from ten other institutions across five countries, all connected via the International Society of Glomerular Disease.
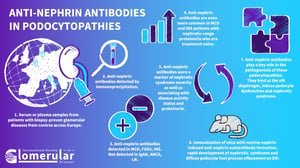
Nephrin is a key adhesion and signaling protein of the slit diaphragm between podocyte foot processes. Its extracellular part protrudes into the space between podocyte foot processes, where it interacts with other molecules and conveys signals to the podocyte through its transmembrane and intracellular part. Changes in nephrin phosphorylation signal to the podocyte via a multitude of adaptor proteins and downstream signaling pathways and result in substantial cytoskeletal reorganization of the podocyte. Nephrin thereby regulates the form and function of podocytes, making it a key protein of the glomerular filtration process. Cases of nephrin disturbance, for example due to genetic mutations in congenital nephrotic syndrome of the Finnish type or in animal models, clearly illustrate its pivotal role for podocyte function. Nephrin dysfunction leads to the deterioration of glomerular filtration with extensive loss of protein into the urine and massive nephrotic syndrome.
Anti-nephrin autoantibodies are antibodies against the key podocyte slit diaphragm protein nephrin. We detected them in about two-thirds of adult patients with minimal change disease and active nephrotic syndrome before the initiation of immunosuppression, and in 90% of children with idiopathic nephrotic syndrome and active nephrotic syndrome before the initiation of immunosuppression. A smaller portion of patients with primary FSGS also exhibited anti-nephrin autoantibodies. In anti-nephrin positive patients, anti-nephrin autoantibodies correlate with clinical disease activity and disease course during onset, remission and relapse of nephrotic syndrome. In a mouse model of active immunization with murine nephrin, animals developed anti-nephrin autoantibodies, which was followed by changes in nephrin phosphorylation, altered nephrin downstream signaling, and the onset of a rapid nephrotic syndrome with a minimal change disease-like histological appearance. From these findings we concluded that anti-nephrin autoantibodies are one of the permeability factors causing nephrotic syndrome in children and adults with idiopathic nephrotic syndrome, minimal change disease, and primary FSGS.
The discovery of anti-nephrin autoantibodies and demonstration of their role thoroughly changes our understanding of associated types of idiopathic nephrotic syndrome, minimal change disease and primary FSGS, which we classify as anti-nephrin associated podocytopathies. It offers an explanation to affected individuals so they can better understand the nature of their disease, rather than a vague and frustrating “idiopathic” label. In the long run and depending on future studies, anti-nephrin autoantibodies might help to predict patients’ risk for different disease courses, guide therapy decisions, and ultimately even set the ground for the development of specific antibody-targeted therapies to avoid the dangerous side effects of the broad immunosuppression that is the current standard of care.
The first signs of a pathological role of antibodies against nephrin in an animal model date back as early as into the late 1980s. Since then, anti-nephrin antibodies have been a matter of debate on different occasions (for example in the context of diabetes and recurrence of nephrotic syndrome after kidney transplantation), reaching a new high with Astrid Weins’ discovery of anti-nephrin autoantibodies in patients with minimal change disease in 2022.
The detection of anti-nephrin antibodies, however, has proven challenging, fostering further discussion on the pathological relevance of anti-nephrin antibodies. The establishment of a reliable and sensitive assay for detection and quantification, its application to large cohorts of international origin and different glomerular diseases, the correlation to clinical disease activity and the creation of an animal model of anti-nephrin-mediated podocytopathy were therefore necessary to fully elucidate the role of anti-nephrin autoantibodies in glomerular diseases.
Of course, the answer to this question is highly speculative. However, the pathobiological understanding of a disease sets the ground for the development of therapeutics which specifically inhibit or counteract underlying pathomechanisms. In the case of anti-nephrin associated podocytopathies, the detection of anti-nephrin antibodies could help us to choose the right patients for antibody-targeting therapies. The current trend towards treatment with anti-CD20 could enhance and/or develop further towards anti-CD38 antibodies in severe cases. In the future, we envision directly targeting autoantibody-producing cells, which was recently demonstrated as a promising therapeutic approach in PLA2R- and THSD7A-mediated membranous nephropathy. Other approaches such as the specific elimination of pathogenic antibodies, inhibition of their binding to nephrin, or drugs interfering with dysfunctional nephrin signaling are also promising options for future investigation.
- First and foremost: How and where do these autoantibodies arise?
- Can we influence or even prevent the onset of anti-nephrin autoimmunity?
- Which factors influence the individual disease course and determine the long-term patient outcome?
- What are the underlying factors in anti-nephrin antibody negative patients?
- What are the molecular consequences of autoantibody binding to nephrin and how can we therapeutically intervene?
Depending on further (prospective) trials, anti-nephrin antibodies as a disease activity marker could serve several different purposes. First, measuring anti-nephrin antibodies during active nephrotic syndrome could help to diagnose anti-nephrin associated podocytopathy in patients who are ineligible for biopsy, for example due to young age, established anticoagulation medication, or pregnancy. This is particularly important since idiopathic nephrotic syndrome and minimal change disease often affect young patients and many patients with nephrotic syndrome are on anticoagulants.
Second, the assessment of anti-nephrin as a prognostic marker in patients with idiopathic nephrotic syndrome, minimal change disease, and focal segmental glomerulosclerosis will be of high interest. Prospective trials are necessary to determine if anti-nephrin antibodies can be used to predict a patient’s response to therapy, disease course, risk of relapse, and so on.
For patients awaiting a kidney transplant, the measurement of anti-nephrin autoantibodies could become an essential part of the pre-transplant workup to reduce the risk of post-transplant recurrence of nephrotic syndrome. During post-transplant follow-up of affected patients, the measurement of anti-nephrin autoantibodies could help to discriminate between recurrence of anti-nephrin associated podocytopathy and other proteinuric disease of the transplanted kidney. A recent study from Japan found a surprisingly high proportion of anti-nephrin positivity in recurrent FSGS patients (Shirai et al., Kidney Int 2024). However, we believe that the role of anti-nephrin antibodies in recurrent disease after transplantation needs further studies.
Collaboration is key! We are deeply grateful for the trust and support of our collaboration partners across the globe, which were enabled by ISGD! ISGD now represents a hub catalyzing collaborative efforts and joint networks as demonstrated by this work.
Further studies are necessary to understand the characteristics and outcomes of patients with anti-nephrin positive podocytopathies, also in comparison to anti-nephrin negative disease. However, the presence of pathogenic antibodies argues for B cell or plasma cell-targeted treatments, aiming at elimination of the pathogenic factor. We envision that response to treatment may be monitored not only based on disease activity (i.e. proteinuria), but also immunological response (i.e. anti-nephrin levels). It would be very valuable to prospectively evaluate new generation monoclonal antibodies targeting B cells, such as obinituzumab and ofatumumab, for their efficacy in reducing anti-nephrin autoantibodies and proteinuria.
The immediate effect of the identification of anti-nephrin antibodies as specific disease markers that strongly correlate with disease activity is that we for the first time have a blood biomarker at hand, allowing to make a specific and pathobiology-based diagnosis. However, the assays to detect this antibody are time-consuming and expensive and it will take time until anti-nephrin antibody measurement becomes routinely available. In the future, we believe that anti-nephrin antibody measurement will allow us to make a noninvasive diagnosis, to guide the treatment of affected patients, and to help to reduce disease recurrence in patients after kidney transplantation.
Currently, this is a purely academic test, developed in the lab at the University of Hamburg. We expect diagnostic companies to develop assays that may be more broadly available. This process takes time, on average a few years. Bringing an assay from the research setting to commercial availability is a complex process requiring:
- Technology transfer and scientific / engineering development
- Clinical trials to assess safety and efficacy
- Regulatory approval process with FDA, EMA and other regulators
- Insurance / health system coverage determinations
Until follow-up prospective studies have been conducted, we won’t have a definitive answer to this question. However, there are B cell-targeted therapies available which may have an impact on anti-nephrin antibodies. In our study, we present the history of three patients with high anti-nephrin antibody levels that were treated with rituximab. This led to a drop in anti-nephrin antibodies and proteinuria in all three cases. One of the three cases had a two-year follow-up after rituximab treatment and their disease did not relapse during this time (in contrast to several relapses before receiving rituximab). We currently use rituximab for relapsing or frequently relapsing disease, and when patients do not respond to glucocorticoids at initial diagnosis.
If patients are refractory to rituximab, this could mean that CD20-negative immune cells are the main disease drivers. These could, for example, be plasma cells which escape the action of rituximab. In this direction, a recent study described considerable success of combined B cell and plasma cell depletion using both rituximab and daratumumab in multidrug-dependent and multidrug-resistant nephrotic syndrome (Angeletti A et al., Am J Transplant 2024).
We are convinced that the knowledge on ANAAs and their pathogenic role in podocytopathies will catalyze the development of compounds specifically targeting the antigen, the antibodies, and the antibody-producing cells.
It is currently too early to say that the ability to detect anti-nephrin antibodies should change biopsy practices in patients with nephrotic syndrome. However, we think that the detection of anti-PLA2R antibodies in membranous nephropathy is a good example of how autoantibody detection can guide further diagnostic steps. The current KDIGO guidelines recommend to not perform a kidney biopsy if anti-PLA2R is present, kidney function is normal and stable, and no other findings such as positive ANA are present. We envision that the ability to detect anti-nephrin autoantibodies might have similar value in the future.
Adult patients were included in our minimal change disease cohort if they had histologically proven minimal change disease lesions as determined by the local nephropathologist. In the presence of any histological signs of beginning capsule synechia or sclerosis patients were classified as Focal Segmental Glomerulosclerosis, which was further subclassified into primary and non-primary depending on diffuse foot process effacement and/or nephrotic-range proteinuria (urinary albumin-to-creatinine ratio (UACR) or urinary protein-to-creatinine ratio (UPCR) > 3.5 g/g) with hypalbuminemia (serum albumin < 3 g/dl). Children were diagnosed with idiopathic nephrotic syndrome by clinical phenotype and laboratory findings, independent of potentially undertaken kidney biopsy.
Apart from potentially a few false-negative patients in our measurements, we expect a separate etiology in anti-nephrin negative patients. Most obviously, one could think of antibodies targeting distinct podocyte proteins, but other as yet unknown permeability factors could also play a role. It is our scientific duty to dig deeper and identify the underlying cause in anti-nephrin negative patients, too.
In our opinion, diseases should be classified based on their endotype, e.g., their pathogenic cause. Therefore, anti-nephrin positive MCD, FSGS or INS should be classified as anti-nephrin associated podocytopathies with (or, in case of INS, without the knowledge of) a MCD or FSGS histotype. However, there may be additional disease factors playing a role, in anti-nephrin positive as well as negative patients. It will be essential to better understand which factors are responsible for the histological as well as clinical phenotype, particularly in terms of the clinical disease course. We need to elucidate which patients with anti-nephrin associated podocytopathy are likely to respond to which therapy and which are at risk for acute or chronic kidney failure.
In our adult cohorts, ethnicity was unfortunately not reported by supplying biobanks. However, people of color were most likely underrepresented in this study due to limited sample availability in supplying (European) biobanks. In our pediatric patients, we had ethnic information for 142 individuals. They comprised 23 Asian, 26 sub-Saharan African, 24 Middle Eastern/North African, 60 Caucasian, and 9 mixed ethnicity pediatric patients, selected due to sample availability in supplying (European) biobanks. Due to the limited data, we are not able to comment on differences across populations. This could be an interesting area for follow-up studies.
Any comments are purely speculative in nature, since we did not assess the origin of measured anti-nephrin autoantibodies in our study. However, the occurrence of idiopathic nephrotic syndrome in children has been linked to upper respiratory tract infections, which could potentially trigger anti-nephrin autoimmunity through unknown mechanisms. Interestingly, we found one child in our control cohort positive for anti-nephrin autoantibodies – and this child had almost 4 times as many infections during the year before sampling compared to the average of all other pediatric controls.
While the pathogenic effect of autoantibodies in podocytopathies known so far mostly depends on traditional immune responses, the detrimental effect of anti-nephrin autoantibodies is most likely due to nephrin dysfunction upon antibody binding. Although the observed change in phosphorylation offers interesting pathomechanistic explanations, the exact mechanism causing nephrin dysfunction upon antibody binding remains elusive so far. The absence of any signs of traditional pathological antibody effects mediated by immune cells or the complement system argues for anti-nephrin associated podocytopathies as a new classification of antibody-mediated podocytopathies.
Outside of podocytopathies, there are other examples of antibody-induced damage due to the interference with the antigen’s physiological functions instead of traditional immune response. For example, autoantibodies in myasthenia gravis impact neuromuscular junction function via different mechanisms including cross-linking of acetylcholine receptors and triggering their endocytosis, interfering with receptor activation by acetylcholine (the classical effect of a receptor antagonist) or by inhibiting acetylcholine receptor clustering through interaction with muscle-specific kinase.
All of the above! We welcome collaboration with additional centers for future studies, and we especially emphasize the need for prospective studies so these findings can be translated into clinical practice to improve patients’ kidney health and quality of life.